
Inherited deficiencies of all of the coagulation factors occur. However, the three most frequent are factor VIII deficiency (hemophilia A or von Willebrand’s disease), factor IX deficiency (hemophilia B or Christmas disease), and factor XI deficiency. Hemophilia A and hemophilia B are inherited as sex-linked recessive disorders with males being affected almost exclusively. The clinical severity of hemophilia A and hemophilia B depends on the measurable level of factor VIII or factor IX in the patient’s plasma. Plasma factor levels less than 1% of normal are considered severe disease, factor levels between 1% and 5% moderately severe disease, and levels between 5% and 30% mild disease. Patients with severe hemophilia have spontaneous bleeds, frequently into joints, leading to crippling arthropathies. Intracranial bleeding, intramuscular hematomas, retroperitoneal hematomas, and gastrointestinal, genitourinary, and retropharyngeal bleeding are added clinical sequelae seen with severe disease. Patients with moderately severe hemophilia have less spontaneous bleeding but are likely to bleed severely after trauma or surgery. Mild hemophiliacs do not bleed spontaneously and have only minor bleeding after major trauma or surgery. Since platelet function is normal in hemophiliacs, patients may not bleed immediately after an injury or minor surgery as they have a normal response with platelet activation and formation of a platelet plug. At times, the diagnosis of hemophilia is not made in these patients until after their first minor procedure (e.g., tooth extraction or tonsillectomy).
Patients with hemophilia A or B are treated with factor VIII or factor IX concentrate, respectively. Recombinant factor VIII is strongly recommended for patients not treated previously and is generally recommended for patients who are both human immunodeficiency virus (HIV) and hepatitis C virus (HCV) seronegative. For factor IX replacement, the preferred products are recombinant or high-purity factor IX. In general, activity levels should be restored to 30% to 40% for mild hemorrhage, 50% for severe bleeding, and 80% to 100% for life-threatening bleeding. Up to 20% of hemophiliacs with factor VIII deficiency develop inhibitors that can neutralize FVIII. For patients with low titers, inhibitors can be overcome with higher doses of factor VIII. For patients with high titer inhibitors, alternate treatments should be used and may include porcine factor VIII, prothrombin complex concentrates, activated prothrombin complex concentrates, or recombinant factor VIIa. For patients undergoing elective surgical procedures, a multidisciplinary approach with preoperative planning and replacement is recommended.
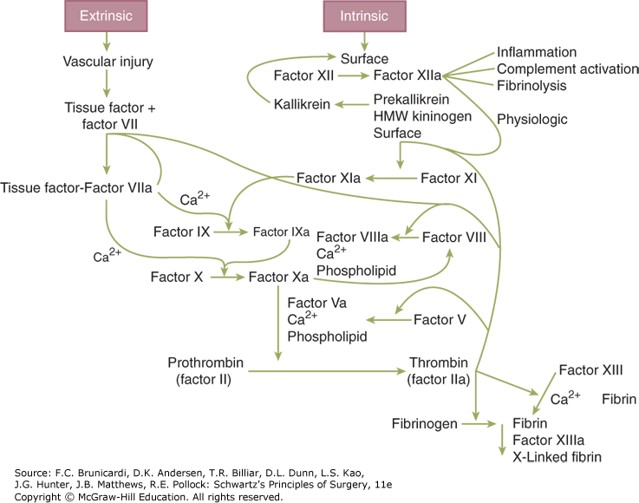
Figure legend: Schematic of the coagulation system. HMW = high molecular weight.
Board Review Questions
1. Which of the following clotting factors is the first factor common to both intrinsic and extrinsic pathways?
A. Factor I (fibrinogen)
B. Factor IX (Christmas factor)
C. Factor X (Stuart-Prower factor)
D. Factor XI (plasma thromboplasma antecedent)
2. Which congenital factor deficiency is associated with delayed bleeding after initial hemostasis?
A. Factor VII
B. Factor IX
C. Factor XI
D. Factor XIII
3. After tissue injury, the first step in coagulation is
A. Binding of factor XII to subendothelial collagen
B. Cleavage of factor XI to active factor IX
C. Complexing of factor IX with factor VIII in the presence of ionized calcium conversion of prothrombin to thrombin
D. Formation of fibrin from fibrinogen
Answers
1. The correct answer is C. Factor X (Stuart-Prower factor)
2. The correct answer is D. Factor XIII
3. The correct answer is A. Binding of factor XII to subendothelial collagen

Create a Free MyAccess Profile
AccessMedicine Network is the place to keep up on new releases for the Access products, get short form didactic content, read up on practice impacting highlights, and watch video featuring authors of your favorite books in medicine. Create a MyAccess profile and follow our contributors to stay informed via email updates.